 Philoclopedia
Philoclopedia
Zusammenfassung: Der Begriff „Junk-DNA“ (z. Dt.: DNA-Müll) scheint notorisch Verwirrung zu stiften. Die populärsten Irrtümer lauten in diesem Zusammenhang: (1) Junk-DNA habe per se keine wie auch immer geartete Funktion. (2) Das Junk-DNA-Konzept sei überholt; Studien wie das ENCODE-Projekt hätten gezeigt, dass 80% der genomischen DNA eine Funktion habe. (3) Die Evolutionstheorie mache über das Auftreten völlig funktionsloser DNA falsche Vorhersagen, könne also die neueren Befunde nicht erklären. (4) Intelligent Design dagegen sage die Existenz von Funktionen in Junk-DNA voraus, liefere also die bessere Erklärung. Im vorliegenden Beitrag werden solche Behauptungen entkräftet. Die Evolutionsbiologie hat nie behauptet, alle Junk-DNA sei völlig funktionslos. Der Funktionsbegriff ist in der Biologie vielschichtig; so kann man Junk-DNA z.B. als DNA verstehen, die keine sequenzabhängige (!) Funktion hat. Barbara MCCLINTOCK nahm bereits 1948 an, dass Transposons regulatorische Elemente beinhalten können. Zudem beruhen die Resultate des ENCODE-Projekts auf fragwürdigen Voraussetzungen: Die These, 80% des Kerngenoms habe eine Funktion, ist nicht haltbar. Für die Entbehrlichkeit eines Großteils nicht-kodierender DNA spricht eine Reihe von Studien; die Pflanze Utricularia gibba beispielsweise hat den Großteil dessen eliminiert, was normalerweise das Genom von Pflanzen ausmacht. Intelligent Design profitiert von ENCODE am wenigsten, und seine Vertreter sind sich selbst nicht einig, ob und wieviel „Funktion“ man zu erwarten habe.
1. Einführung
Biologische Merkmale, die keine essenziellen Aufgaben (mehr) erfüllen, sprechen oft in besonderer Weise für einen nicht intendierten, evolutionären Entstehungsprozess. So weisen die Evolutionsbiologen darauf hin, dass die „Funktionslosigkeit“ biologischer Strukturen auf die gemeinsame Abstammung der Arten hindeute und darüber hinaus oftmals auch etwas über die Mechanismen ihrer Entstehung verrate. Dem halten vor allem Evolutionsgegner (z.B. die Verfechter des „intelligenten Designs“) entgegen, ein Merkmal würde nicht aus purer Traditionsliebe Jahrmillionen überdauern, sondern weil es konstruktiv notwendig sei. In diesem Zusammenhang glaubt man nachgewiesen zu haben, dass sich die vermeintlichen Paradebeispiele funktionsloser Merkmale im Zuge der Forschung regelmäßig in Luft auflösen, wodurch wiederum eine intelligente „Planung“ an Plausibilität gewönne.
Insbesondere, was die sog. „Junk-DNA“ (zu Deutsch: Erbgut-Gerümpel) anbelangt, erhitzt die Debatte, inwieweit es sich tatsächlich um überflüssige bzw. funktionslose DNA handelt, regelmäßig die Gemüter. Während Genetiker große Teile des Genoms als funktionslos ansehen, behaupten Anhänger des Intelligent Design das Gegenteil und verbuchen den tatsächlichen oder angeblichen Nachweis funktionaler Anteile in „Junk-DNA“-Abschnitten als Bestätigung ihres Design-Ansatzes.
Die Analyse der Genome lebender Organismen läuft nun schon seit drei Jahr zehnten, und es ist aus mehreren Gründen schwierig, in ihnen die funktionalen Elemente – die „Information“ – zu lokalisieren. Dafür gibt es drei Gründe:
- Immer wieder herrscht Konfusion um den „Sinn“ und die „Funktion“ biologischer Strukturen – seien es Gene, anatomische Organe oder Verhaltensweisen. Eine nähere Betrachtung zeigt, dass in solchen Fällen oft begriffliche Missverständnisse vorliegen, wenn nämlich menschliches Denken auf Naturprozesse wie die Evolution übertragen wird (MAHNER 2001; MAHNER/ BUNGE 2001).
- Der Begriff der Funktion selbst ist mehrdeutig und wird in mehr als einer Bedeutung verwendet (MAHNER 2001; MAHNER/BUNGE 2001). Je nach Definition kann es also sein, dass die betreffenden Strukturen funktional sind oder nichtfunktional.
- Die wissenschaftliche Lokalisierung und Zuordnung „genetischer Information“ zu Genomteilen ist weit schwieriger als sich ein Laie vorstellen kann.
Dieser Beitrag soll Lichts ins Dunkel der Junk-Debatte bringen und zu allererst die Begrifflichkeiten klären. Zunächst stellen wir ein paar Überlegungen zu den Begriffen Sinn und Funktion an. Anschließend erörtern wir, was man (sinnvollerweise) unter „Junk-DNA“ versteht und was (entgegen populären Irrtümern) nicht darunter verstanden werden kann. Wir werden erläutern, welche Sorten von Junk-DNA es gibt und diskutieren, inwieweit das Vorliegen von Junk-DNA die Evolutionstheorie belegt. Abschließend nehmen wir auf „Intelligent Design“ (ID) Bezug und erörtern, warum - entgegen der engagierten Behauptungen ihrer Vertreter - das Vorliegen von teils funktionaler (Junk-) DNA weder von „Intelligent Design“ vorausgesagt wurde, noch vorausgesagt werden kann.
2. Freund oder Feind?
Um sich dem Thema Junk-DNA zu nähern, muss man sich zuallererst über den Begriff der biologischen Funktion(alität) Klarheit verschaffen. Dabei ist es äußerst wichtig, sich von menschlichem Denken frei zu machen. Dies lässt sich sehr gut an ein paar Beispielen erläutern: Bekanntermaßen stehen Organismen z.B. in einem symbiotischem Verhältnis oder in einem Räuber-Beute Verhältnis. Nach menschlichem Maßstab schließt sich beides gegenseitig aus. Aber die Biologie (genauer: die Ökologie) denkt nicht und beabsichtigt nichts, daher wird man vieles nicht verstehen, wenn man menschliche Denkstrukturen in die Biologie projiziert. In diesem Abschnitt werden wir am Beispiel „Freund oder Feind“ (Räuber Beute oder symbiotische Kooperation?) sehen, dass solches menschliche Schubladen-Denken zu kurz greift und werden diese Erkenntnis auf die Frage „JunkDNA – funktional, schädlich oder unnütz?“ übertragen können.
Die Geschichte des Lebens auf diesem Planeten kann man in Bezug auf Komplexität mit einem Schachspiel vergleichen – die Gemeinsamkeit ist folgende: Die Grundprinzipien der Evolution sind so einfach, dass man ihre Regeln bzw. Mechanismen auf zwei Schreibmaschinenseiten zusammenfassen kann. Durch die Wechselwirkungen im System wird das Ganze aber so komplex, dass über beide Themen ganze Bibliotheken geschrieben worden sind. Die Folge ist, dass die Kenntnis eben dieser Grundprinzipien noch längst nicht ausreicht, um das Ganze wirklich zu verstehen – und das gilt für das Schachspiel ebenso wie für den Evolutionsprozess. Im Bereich der Evolution kommt erschwerend hinzu, dass die Verwendung von Begriffen der Umgangssprache („Sinn“, „Zweck“, „Schaden“, „Kampf ums Dasein“) in vielen Fällen Verwirrung stiftet, weil damit menschliche Attitüden in einen Naturprozess hinein projiziert werden. So führt die Wechselwirkung zwischen Organismen, Arten, Populationen dazu, dass sie beispielsweise in einem Räuber-Beute-Verhältnis stehen (oder vergleichbar in einem Wirts-Parasiten-Verhältnis), oder dass sie sich gegenseitig verdrängen (ökologische Konkurrenz) oder aber wechselseitig aufeinander angewiesen sind (z.B. in Form von Symbiosen). Da liegt es nahe, die betreffenden Arten als „Feinde“ oder „Kooperationspartner“ zu betrachten, und bekanntlich ist man im menschlichen Leben das eine oder das andere. Aber schauen wir uns einmal zwei Beispiele an:
- Weidevieh (Rinder, Antilopen etc.) ernährt sich von Steppengras, das ist vom Prinzip her ein klassisches Räuber-Beute-Verhältnis. Konsequenterweise hat eine Koevolution – in diesem Zusammenhang so etwas wie ein „evolutionäres Wettrüsten“ – stattgefunden: Gräser haben Bitterstoffe entwickelt und schützen sich ferner durch harte und scharfe SilikatKristalle auf der Blattoberfläche so gut wie möglich vor Fraß: Fast jeder dürfte sich schon einmal an Gras geschnitten haben. Im Gegenzug sind die Zungen und Zähne der Weidetiere immer härter geworden. Andererseits sind Gräser jedoch auf Beweidung angewiesen, denn ohne Weidetiere würden Büsche und Bäume sehr schnell die Graslandschaften überwachsen und dem Gras das Licht nehmen. Das trägt Züge einer typischen Symbiose: Gräser versorgen Rinder mit Nahrung und werden im Gegenzug dafür gegen Überwuchs geschützt. Im welchem Verhältnis also stehen Rinder und Gräser denn nun – in einem Räuber-Beute- oder in einem symbiontischen Verhältnis? Feind oder Freund? Antwort: Es ist beides gleichzeitig!
- Der menschliche Organismus beherbergt mehr Bakterien als er selbst Körperzellen besitzt. Zum einen versorgen uns Darmbakterien mit Vitamin K, die Hautflora schafft den schützenden Säuremantel usw. Dafür stellen wir ihnen einen Lebensraum und Nahrung zur Verfügung – eine klassische Symbiose. Andererseits hat unser Organismus ein riesiges Arsenal an Mechanismen entwickelt, um Bakterien –auch die symbiontischen! – abzuwehren und in Schach zu halten. Wenn diese Mechanismen versagen, so wie z.B. bei AIDS, also im Endstadium einer HIV-Infektion, werden diese symbiontischen Bakterien zu einer tödlichen Gefahr. Also sind sie unsere Freunde oder Feinde? Wiederum: beides gleichzeitig! (KHOSRAVI/MAZMANIAN 2013; s. auch das NIH Human Microbiome Project; The International Human Microbiome Consortium).
Die hier angedeuteten Wechselwirkungen sind im Rahmen der Evolutionstheorie ohne Schwierigkeit beschreibbar, sie können sogar in mathematischen Modellen beschrieben werden (MCNAMARA 2013; NUISMER et al. 2013; SUWEIS et al. 2013 und die dort zitierten Arbeiten). Problematisch wird es erst dann, wenn man versucht, solche Systeme in Alltagssprache zu beschreiben. Die Evolution ist ein natürlicher Prozess, der keinen Willen und keine Absicht, keine Freunde und keine Feinde, keinen Sinn und keine Bedeutung im menschlichen Sinne kennt. Daher ist es möglich, dass eine Wechselwirkung gleichzeitig vorteilhafte und nachteilige Aspekte (i.S. der Auswirkung auf den Fortpflanzungserfolg) haben kann – und das ist etwas, was sich umgangssprachlich nur schwer fassen lässt. Dies ist eine Quelle schier unausrottbarer Missverständnisse, wenn es um die populärwissenschaftliche Darstellung der Evolution geht.
3. Funktional oder nutzlos? - Primäre und sekundäre Funktionen
Ähnliche Probleme ergeben sich, wenn man in der Alltagssprache die Funktionalität von biologischen Strukturen beschreiben möchte. „Funktion“ impliziert umgangssprachlich einen Plan und eine Absicht bzw. einen konkreten, vorgegebenen Zweck. Die in der Biologie vielleicht wichtigste Definition des Begriffs Funktion ist dagegen, bezogen auf die Evolutionstheorie, mit der differenziellen Tauglichkeit seines Besitzers assoziiert (vgl. MAHNER/BUNGE 2000). Man kann sagen, ein Merkmal erfülle genau dann eine Funktion, wenn es seinem Besitzer einen Selektionsvorteil im ökologischen Kontext gegenüber den Nicht-Besitzern beschert. Das bedeutet, dass sich der Besitz des betreffenden Merkmals auf das Leben und die Fortpflanzungsfähigkeit ihrer Besitzer auswirken. Daher kann ein „Ingenieurs-Blick“ auf biologische Strukturen zu Verwirrungen führen. Auch hierzu einige Beispiele:
- Flugfähige Insekten haben zwei Flügelpaare, ideal zu sehen bei Libellen. Bei den Dipteren (Fliegen) sind die Hinterflügel zu sog. Halteren (gesprochen: „Halteeren“) verkümmert; sie tragen zum Flug nichts mehr bei. Stattdessen schwingen sie im Takt der Flügel mit, und wie man mittlerweile weiß, dienen sie dem Insekt als Trägheitskompass zur Navigation (KLOWDEN 2007). Sind sie nun „nutzlose Flügel“ oder „funktionale Strukturen“? Nun, beides! Denn auch hier gilt: Die Flügel haben ihre primäre Funktion eingebüßt, aber eine sekundäre Funktion gewonnen.
- Verfolgt man makroevolutive Umbildungen in der Stammesgeschichte und somit Veränderungen über sehr lange Zeiten, erkennt man unschwer, dass Strukturen, die funktionslos geworden sind, zurückgebildet werden. So haben Landwirbeltiere keine Kiemen mehr, Schlangen keine Beine und Wale keine Zähne und Extremitäten. Es fällt aber auch auf, dass viele dieser Strukturen bzw. deren Anlagen in der Embryonalentwicklung noch durchlaufen (rekapituliert) werden und dass Reste (Rudimente) von ihnen sogar in der Adultform (beim ausgewachsenen Tier) noch vorhanden sind. Sind sie nun „nutzlose Reste der Evolution“, oder „tragen sie eine neue Funktion“ oder „sind sie wichtig für die Körperentwicklung“? Und wiederum: all dies schließt sich nicht gegenseitig aus!
Das Problem ist hier offenbar ganz ähnlich wie oben am Beispiel des Weideviehs diskutiert: Umgangssprache und Ingenieursblick sind schlechte Ratgeber, um diese Verhältnisse zu beschreiben. Man muss sich, um den Evolutionsprozess zu verstehen, von menschlich-zweckgerichtetem Denken (Teleologie: „Sinn“, „Ziel“, Absicht“ usw.) frei machen: Der Evolutionsprozess kann nur das erfassen, was vorhanden ist und was sich in kleinen oder mittleren Schritten ändern oder neu kombinieren lässt. Ein Organismus kann nicht „wegen Umbau für ein paar Tausend Generationen schließen“ – es muss eine kontinuierliche Entwicklung mit kleinsten, kleinen und hin und wieder etwas größeren Entwicklungsschritten geben. Das hat zur Folge, dass einmal vorhandene Strukturen nur selten wieder komplett verschwinden.
Anders gesagt kann die Evolution nicht einfach „etwas Nötiges oder Sinnvolles erfinden“ – sie greift an vorhandenen Strukturen an, es kann gar nicht anders sein. Stellen wir uns folgendes Szenario vor: Aufgrund der Biomechanik des Fliegenkörpers haben sich die Vorderflügel so entwickelt, dass sie die ganze Arbeit leisten. Damit werden die Hinterflügel funktionell verzichtbar; ihr Verlust wäre nicht nachteilig. Nun sind Mutationen, die auf einen Schlag die komplette Rückbildung, das gänzliche Verschwinden einer anatomischen Struktur verursachen, extrem selten (und oftmals aufgrund der komplexen Verflechtungen der entwicklungsbiologischen Steuerungssysteme auch gar nicht möglich!); ergo wird man erwarten, dass die Hinterflügel langsam verkümmern. Indem sie nun frei von Selektionsdruck sind, können sie schlussendlich verschwinden oder aber sie übernehmen eine neue Funktion: Die verkrüppelten, verkleinerten Hinterflügel haben eine höhere Trägheit bei einem verschwindenden Luftwiderstand, verhalten sich also wie ein Kreisel(kompass). Mit diesen veränderten physikalischen Parametern erfahren sie einen neuen Selektionsdruck, eben in Richtung der Optimierung eines Navigationssystems.
Ein weiterer Aspekt: Technische Strukturen (Maschinen, Gebäude etc.) werden einzeln und in Teilen erschaffen und dann zusammengebaut. Biologische Strukturen hingegen entwickeln sich in der Embryogenese in einem ganzen, zu jedem Zeitpunkt voll funktionsfähigen System. Die Signale zur Ausbildung anatomischer Strukturen werden von anderen Strukturen gegeben, z.B. gibt der Urdarm Signale zur späteren Ausbildung der Lunge, die Zahnanlagen Signale zur Formbildung des Kiefers etc. Wenn also Strukturen wie die Kiemenbögen unserer FischVorfahren physiologisch funktionslos werden, so sind diese Kiemenbögen (genauer: die Visceralbögen) dennoch bei Landwirbeltieren in die embryologischen Bildungsprozesse eingebunden, so dass von dorther sehr wohl noch ein starker Selektionsdruck auf ihre Erhaltung gegeben ist – obwohl sie ihre ursprüngliche Funktion längst verloren haben (Überblick in BOMMAS-EBERT et al 2006). Analoges gilt für die Zahnanlagen, die bei den Bartenwalen zurückgebildet, während ihrer Embryogenese jedoch eisern rekapituliert werden. Der Grund ist, dass die Vorfahren der Bartenwale Zähne besaßen und die Zahnanlagen bei der Formbildung des Kiefers beteiligt waren. Folglich können die Zahnanlagen nicht mehr nachträglich eliminiert werden, wenn eine formgerechte Ausbildung des Kiefers gewährleistet bleiben soll.[1]
Man müsste also sagen: Im Sinne der primären Funktion sind die Halteren bei Insekten, die Kiemenbögen bei Säugern sowie die Zahnanlagen bei Bartenwalen nutzlos geworden. Sie erfüllen jedoch andere (d.h. sekundäre) Funktionen: Die Halteren als Navigationsorgane und die Visceralbögen und Zahnanlagen als Signalgeber in der Keimesentwicklung. Damit ist die Frage „nutzloses Rudiment oder funktionales Organ?“ nur zu beantworten mit: „beides!“.
Der Kritiker könnte hier einwenden, die Identifikation von Primärfunktionen sei nicht ohne weiteres möglich, sondern setze bereits konkrete stammesgeschichtliches Annahmen voraus, so dass es sich gar nicht um einen unabhängigen Beleg für Evolution handele, sondern um einen Zirkelschluss: Das, was belegt werden soll, nämlich Evolution, werde in Gestalt der Deutung von Primärfunktionen bereits vorausgesetzt. Dem ist aber nicht so, denn mutmaßliche Primärfunktionen lassen sich zunächst auch ohne evolutionäre Annahmen rekonstruieren, sofern die betreffenden Merkmale komplex genug sind, um sie mit den Merkmalen anderer Arten, welche die betreffende Primärfunktion noch aufweisen, in Beziehung zu setzen. So entsprechen die Zahnanlagen der Wale bis ins Detail den Anlagen anderer Säugetiere, bei denen sich diese später zu Kauwerkzeugen entwickeln.
4. Ein Testfall für die Evolutionstheorie
Gerne werden technische Systeme mit Lebewesen verglichen – insbesondere in der populärwissenschaftlichen Literatur. Interessant sind allerdings dabei weniger die Ähnlichkeiten – sie erklären sich aus dem Selektionsdruck auf physiologische Funktionalität, der oftmals ähnliche Strukturen hervor bringt wie ingenieurtechnische Entwicklungsarbeit. Bemerkenswert und aufschlussreich sind jedoch vor allem die Unterschiede, die auf der grundsätzlichen Verschiedenheit zwischen einem geplanten Design (Technik) und einem ungeplanten Naturprozess (Evolution, rückgekoppelt über den Fortpflanzungserfolg) beruhen.
In einem kreativen Design-Prozess sind Design, Materialauswahl, Produktion und Zusammenbau weitestgehend getrennt. Daher ist es möglich, ähnliche oder faktisch identische Produkte alternativ aus ganz unterschiedlichen Materialien herzustellen. Im Evolutionsprozess kann man hingegen nicht erwarten, dass z.B. bei Vögeln das verhältnismäßig schwere Knochenmaterial (Calcium-Apatit) „sinnvollerweise“ durch leichteres Material ersetzt wird, denn die entsprechenden Genwirkketten und ontogenetischen Regelkreise sind im genetischen Gesamtplan des Organismus eng miteinander verknüpft. Wie oben bereits erwähnt könnte ein Designer „vom Reißbrett aus“ Teile austauschen (bei entsprechender Kenntnis des Systems wäre das auch bei Lebewesen möglich), der Evolutionsprozess kann das jedoch nicht bzw. nur eingeschränkt (vgl. BEYER 2007).
Auch kann man bei verschiedenen technischen Apparaten gleiche Teile einbauen; so z.B. finden sich identische Scheibenwischer oder baugleiche Einspritzpumpen von Bosch in ganz unterschiedlichen PKW. Wenn sich hingegen in einer Tiergruppe – z.B. bei Vögeln – Flügel entwickeln, so kann diese Neuerung von anderen Tiergruppen nicht übernommen werden, wie es im technischen Design problemlos möglich ist. Ergo wird man erwarten müssen, dass – wenn die Randbedingungen es erlauben und ein entsprechender Selektionsdruck besteht – parallele Entwicklungen z.B. von Flügeln zu anatomisch gänzlich verschieden aufgebauten Strukturen führen, und in der Tat haben Vogel-, Fledermaus-, und Insektenflügel in Aufbau und Entwicklung nichts miteinander zu tun.
Umgekehrt würde kein Ingenieur auf die Idee kommen, technische Vorrichtungen zur Fortbewegung auf dem Land (Beine) im Wasser (Flossen), in der Luft (Flügel) oder im Boden (Grabeschaufeln) nach demselben Bauprinzip zu entwickeln. In der Evolution hingegen bleibt gar keine andere Möglichkeit: Die Landwirbeltiere haben nun mal nur eine ganz bestimmte Art von Extremitäten (die 5-strahlige Tetrapodenextremität); diese musste sich an besagte Fortbewegungsarten anpassen – und konnten das auch, soweit Art und Aufbau der Extremitäten diese Varianten biomechanisch erlauben (s. BEYER 2007).
Weil in einem Design-Prozess immer wieder von neuem geplant werden kann, gibt es auch keine Notwendigkeit, an sinn- oder funktionslos gewordenen Strukturen festzuhalten. Weil aber (insbesondere bei tierischen) Organismen die Organe und Strukturen in den Prozess der Embryogenese eingebunden sind, können sie in vielen Fällen nicht einfach komplett verschwinden. Jede Neuerung muss auf Vorhandenem aufbauen, was bisweilen absurde Konsequenzen hat. Ein Beispiel: Der rückläufige Kehlkopfnerv (nervus laryngeus recurrens) macht einen Umweg durch den Körper, er läuft vom Hirn zum Herzen und wieder zurück zum Kehlkopf. Bei der Giraffe führt das dazu, dass er für eine Distanz von einigen cm einem Umweg von etlichen Metern (!) macht, ohne dass es unterwegs noch Abzweigungen gäbe o.ä. Der Grund liegt darin, dass unsere Fisch-Vorfahren keinen Hals hatten; somit macht der Nerv bei seinem Weg am Herzen vorbei keinen Umweg. Als sich bei Säugetieren immer längere Hälse entwickelten (wobei die Kiemen längst verschwunden waren), war der Nerv immer noch auf die entsprechende Umgebung angewiesen, um bei seiner Entwicklung im Embryo seinen Weg zu finden. Weil diese Konstellation aus Sicht der primären Funktion zwar völlig unsinnig ist, aber keinen signifikanten Nachteil mit sich bringt, gab es auch keinen Selektionsdruck, diese „Fehlkonstruktion“ zu korrigieren (DAWKINS 2009). Doch selbst wenn ein Selektionsdruck derartiges fordern würde, wäre eine Korrektur aus Sicht der sekundären (embryogenetischen) Funktion heute kaum mehr möglich: Waren die Kiemen und Aortenbögen mit ihrem Geflecht an Blutgefäßen und Nerven nämlich erst einmal im Bauplan der „Fische“ etabliert, musste jede evolutionäre Weiterentwicklung unbedingt die Funktionsfähigkeit des Merkmalsgefüges gewährleisten.
Als die Kiemen später in der Evolution verschwanden und die Landwirbeltiere Hals und Kehlkopf entwickelten, war es aus entwicklungsbiologischer Sicht lediglich möglich, die Aortenbögen zu modifizieren und die Strukturen in ihrer zeitlichen und räumlichen Entwicklung zu verschieben. Es war aber nicht möglich, die Aortenbögen auszurangieren und das komplexe Gewirr aus Blutbahnen, Organen und Nervensträngen grundlegend neu zu ordnen, um zu verhindern, dass beim Verlängern des Halses und beim Abstieg (Deszensus) des Herzens in den Brustkorb der vierte bis sechsten Aortenbogen mit nach unten wandert und dabei den Nervus laryngeus recurrens mitnimmt. Deshalb schlingt sich der Nerv um den Aortenbogen und wurde evolutionär schrittweise, Millimeter um Millimeter, verlängert. Offenbar ist hier der Grenznutzen einer sich in mehreren Schritten vollziehenden, jeweils nur kleinen Verlängerung des Nervs aus selektionstheoretischer Perspektive höher als eine grundlegende Umgestaltung des Bauplans: Mutationen, die den Weg des Kehlkopfnervs „optimieren“ und das Chaos im Brustkorb beseitigen würden, sind zu komplex und zu unwahrscheinlich, weil sie viele Merkmale gleichzeitig ändern müssten, wobei die negativen Folgelasten pleiotroper Mutationen durch weitere Mutationen ausgeglichen werden müssten.
Die primäre Funktion der in der Säugerentwicklung transitorisch auftretenden Aortenbögen ist aufgrund der komplexen Struktur und Ähnlichkeit mit dem entsprechenden „Fischbauplan“ noch erkennbar. Wir haben also keinen Grund, am stammesgeschichtlichen Ursprung dieses (aus Sicht der Sekundärfunktion) dysfunktionalen Gewirrs aus Blutbahnen und Nervensträngen, einschließlich des rückläufigen Kehlkopfnervs, zu zweifeln. Die Evolutionstheorie hat den Testfall bestanden. Aus Ingenieurs- bzw. Designersicht dagegen lässt sich dieses Beispiel nicht erklären, denn die Neuordnung des Blutbahnen- und Nervengeflechts wäre weit effizienter als die ziemlich starre Wiederholung eines alten Bauplans, für den es bei den Säugetieren keine prospektive Bedeutung mehr gibt, weswegen er auch nur transitorisch in Erscheinung tritt.
5. DNA und Gene
Was für Organismen und ihre Wechselbeziehungen, für Funktion und Funktionswandel gilt, das findet seine Entsprechung auch bei genetischen Elementen. Konkret gibt es immer wieder Verwirrung um den Begriff „Junk-DNA“ („Schrott-“ oder „Müll-DNA“). Und im Streit um die Evolutionstheorie erhitzen sich die Gemüter regelmäßig an der vermeintlichen Funktion oder Funktionslosigkeit dieser Junk-DNA. Seit OHNO (1972) versteht man darunter Sequenzen im Genom, die (vermeintlich oder tatsächlich) keine Funktion besitzen. In der Tat kodiert nur ein kleiner Teil des Säuger-Genoms (und somit auch unseres eigenen) für Proteine; es dürften unter 5% sein. Zusätzlich gibt es die nicht-Protein-kodierenden, aber funktionalen RNAs, von denen man tRNA und rRNA schon lange kennt, snoRNA, si/miRNA, XIST-RNA und viele andere sind mittlerweile hinzu gekommen[2]; sie mögen zusammen nochmals einige Prozente des Genoms ausmachen. Zählt man nun regulatorische Elemente (Promotoren, Enhancer / Silencer, Splice-Sites[3] etc.) und strukturelle Bereiche (Zentromer- und Telomer-DNA) hinzu, so bleibt am Ende immer noch ein Anteil von mehr als der Hälfte, der „funktionslos“ ist.
Im Folgenden soll zunächst geklärt werden, was man im Zusammenhang mit „Junk-DNA“ unter „funktionslos“ versteht. Anschließend werden einige Beispiele erörtert wie z.B. die sog. Pseudogene, „tandem repeats“, und neuere Ergebnisse aus dem ENCODE-Projekt (ENCODE Project 2012) diskutiert. Abschließend wird der Frage nachgegangen, ob das Deutungsschema des Intelligent Design über die Funktion und Entstehung dieser DNA-Elemente überhaupt etwas wissenschaftlich Relevantes aussagen kann.
6. Was ist Junk-DNA? Oder: Gibt es funktionslose Gene?
Die Frage, ob es Merkmale bzw. Bereiche im Erbgut der Lebewesen gibt, die keine Funktion erfüllen, ist nicht ganz einfach zu beantworten. Einerseits liegt das an der bekannten Begründungsasymmetrie: Funktionen lassen sich im konkreten Einzelfall nachweisen, schwerlich aber das Fehlen einer Funktion. Übt ein Merkmal keine erkennbare Funktion aus, lässt sich immer behaupten, die Funktion sei einfach noch nicht gefunden worden.
Andererseits hängt die Antwort davon ab, was man konkret unter einer Funktion versteht. Wie bereits erläutert wird im Rahmen evolutionärer Fragestellungen der Funktionsbegriff oft mit einem Selektionsvorteil gleichgesetzt, den ein Merkmal X einem Organismus Y in einer Umwelt Z beschert. Häufig wird der Begriff „Funktion von X“ aber auch gleichgesetzt mit „Aktivität eines Merkmals X“, welches im Organismus Y einen physiologischen Prozess bedingt oder in Gang hält, nicht aber zwangsläufig einen Selektionsvorteil bringt. Z.B. fand man heraus, dass etwa 10 Prozent der Pseudogene transkribiert werden, also im Sinne einer biologischen Aktivität funktional ist. Die Frage, ob diese Transkripte auch einen biologischen Nutzen haben bzw. ihren Besitzern einen Selektionsvorteil bescheren, ist aber eine ganz andere: Viele werden nur zu unnützer Junk-RNA umgeschrieben, in anderen Fällen erfüllen die (nicht mehr Protein-kodierenden) Transkripte dagegen eine (sekundär erworbene!) regulative Funktion.
Es wird noch komplizierter: Lässt sich nachweisen, dass einem biologischen Merkmal eine wie auch immer geartete Funktion zukommt, kann damit noch nichts über den strukturellen Grund dieser Funktion ausgesagt werden – ja, es ist noch nicht einmal klar, ob es sich überhaupt um eine strukturabhängige Funktion handelt, denn nicht alle Strukturen (oder besser: Komponenten eines Merkmals) sind gleichermaßen essenziell für die Funktion. Das heißt, biologische Merkmale können Komponenten aufweisen, die für die Funktion gar nicht (mehr) relevant sind. Beispielsweise die Tatsache, dass die Extremitäten von Fledermaus, Vogel, Maulwurf, Mensch, Wal und Eidechse gleichartig aufgebaut sind: Sie verfügen über die charakteristische Anordnung von Oberarmknochen, Elle, Speiche und Handwurzelknochen, sowie über meist fünfstrahlige Finger. Angesichts dieser funktionalen Verschiedenartigkeit ist offensichtlich, dass der identische Grundbauplan der Extremitäten nicht funktionell begründet ist - kein Ingenieur käme etwa auf die Idee, ein Fahrgestell, eine Flug-, Tauch- und Schwimmapparatur nach gleichem Aufbauschema zu konstruieren.
Was also ist der Grund für die Gleichheiten im Bauplan? Warum ist der Feinbau der Extremitäten so und nicht anders? Antworten darauf liefert die evolutionäre Entwicklungsbiologie. Wie bereits ausgeführt wurde, spiegelt sich hier evolutionäre Geschichte und damit historisch bedingte Organisationsstrukturen (und damit auch Entwicklungszwänge) wieder. Da die Tetrapoden von devonischen Fischen abstammen, war die Evolution „gezwungen“, auf die vorhandenen Ressourcen (die Struktur der ersten Extremitäten) zurückzugreifen. Und da gemäß einem treffenden Bonmot von G. OSCHE die von ihnen abstammenden Arten nicht „wegen Umbau vorübergehend geschlossen“ werden können, blieb die Struktur aus konstruktiven Gründen erhalten.
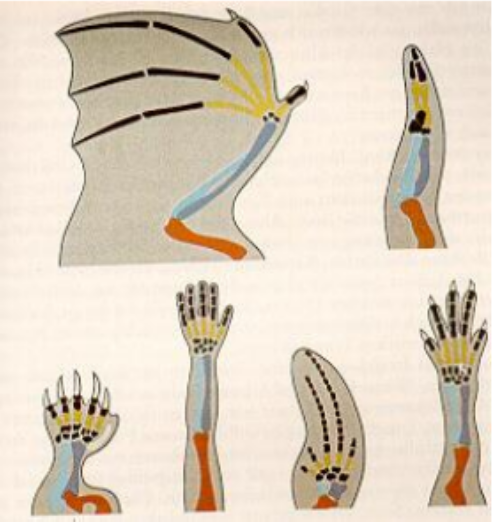
Abbildung: Ähnlichkeiten im Skelettbau der Extremitäten von Vierfüßern. Von oben nach unten und von links nach rechts: Fledermaus, Vogel, Maulwurf, Mensch, Wal und Eidechse. Trotz unterschiedlichster funktioneller Beanspruchung ist der Feinbau der Skelette identisch. Rot: Oberarmknochen, blau: Elle und Speiche, gelb: Mittelhandknochen, schwarz: Handwurzelknochen und Fingerknochen (Phalangen).
Analoge Betrachtungen müssen wir auf genetischer Ebene anstellen: Alle Gene weisen in ihren Sequenzen Anteile auf, die für ihre Funktion wichtig, weniger wichtig und völlig unwichtig sind. Dementsprechend ist auch bei der jedem DNA-Element im Genom sorgfältig zu unterscheiden, (a) ob es an sich eine (strukturbzw. sequenzabhängige) Funktion erfüllt, oder (b) ob der betreffende DNAAbschnitt Teil einer anderen (übergeordneten) Funktionsstruktur (bzw. DNAAbschnitts) ist oder aber (c) ob er tatsächlich völlig funktionslos ist (in dem Sinne, dass er ohne jede Auswirkung Folge deletiert werden könnte). Damit zeigt sich nun, dass die ursprüngliche Definition von „Junk“ zu naiv war: Bei „Funktion“ hatte man an die Sequenz im Blick, folglich war alles andere unfunktional und damit „Müll“. Daher muss man heutzutage die Definition von „Junk“ verfeinern: „jeder DNA-Abschnitt, der keine Sequenz-abhängige Funktion trägt“. Dies bedeutet, dass Junk-DNA immer nicht kodierend ist - damit ist aber längst nicht jede nicht-kodierende Sequenz "funktionslos" oder "sinnlos". Nicht kodierende Abschnitte auf der DNA sind beispielsweise die so genannten Introns. Es handelt sich dabei um nicht-(Protein-) kodierende Bereiche in einem Gen, die nach der Transkription des Gens zu prä-RNA durch „Splicing“ entfernt werden. Sie erfüllen auch eine Funktion, indem sie beispielsweise alternatives Splicing ermöglichen (Abb. 3). Diese Funktion ist aber nicht sequenzabhängig (bis auf die Sequenz an der Splice site und die regulorischen Sequenzen in der Nähe), das heißt das Intron kann experimentell verkürzt oder durch eine andere Sequenz ersetzt werden, ohne die „Fitness“ der Zelle zu beeinträchtigen. Daher wird man nach alter Lesart Introne als "Junk und somit funktionsloser Müll" betrachten, heute muss man jedoch sinnvollerweise sagen: Manche Introne besitzen gar keine Funktion (sie können wegfallen), andere hingegen ermöglichen das differenzielle Spleißen, damit tragen sie eine (Sequenz-unabhängige) Funktion.
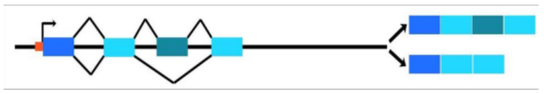
Abbildung: Alternatives Splicing. Die blauen Kästen symbolisieren Exons, das orangefarbene Kästchen den Transkriptionsstartpunkt und die Bereiche zwischen den Exons sind Introns. Bei der Transkription (dem Umschreiben der DNA zur RNA) wird zunächst der gesamte Bereich zwischen Transkriptionsstart- und Endpunkt in prä-RNA umgeschrieben. Aus dieser prä-RNA werden die Introns ausgeschnitten, so dass die Exons direkt aufeinander folgen (d. h. die Exons werden zusammen „gespleißt“). Die resultierende RNA wird dann als Boten- oder Messenger-RNA (mRNA) bezeichnet und erst dann in eine Aminosäuresequenz/Protein translatiert („übersetzt“). Beim alternativen Splicing können aus einer prä-RNA verschiedene mRNAs und damit auch verschiedene Proteine entstehen, indem z. B. wie im Bild ein Exon (der dritte Kasten von links) entweder in die entstehenden mRNA mit eingeht (rechts oben) oder nicht (rechts unten).
Halten wir fest: Der Begriff „funktionslos“ hat verschiedene Bedeutungen. Die Bedeutung im allgemeinen Sinne wäre: gar keine Funktion, ganz gleich welche, Halten wir fest: Der Begriff „funktionslos“ hat verschiedene Bedeutungen. Die Bedeutung im allgemeinen Sinne wäre: gar keine Funktion, ganz gleich welche, also auch keine sequenzunabhängige Funktion wie die für Introns beschriebene. Die Bedeutung im engeren Sinne ist dementsprechend: keine struktur- bzw. keine sequenzabhängige Funktion für den Organismus. Diese Unterscheidung ist eine sehr wichtige, wenn man über Junk-DNA spricht, da hier funktionslos immer im Sinne von „keine sequenzabhängige Funktion“ gemeint ist! Sehen wir uns im Folgenden einige Beispiele von „Junk-DNA“, nämlich Pseudogene und Repeats (wie die sog. „tandem repeats“) unter diesem Aspekt näher an.
7. Beispiele für Junk-DNA
7.1. Pseudogene
Pseudogene sind DNA-Abschnitte, die zwar wie ein Gen aufgebaut sind, die jedoch keine funktionale RNA bzw. kein Protein hervor bringen, weil sich in ihnen Mutationen angesammelt haben, so dass sie ihre primäre Funktion eingebüßt haben. Die meisten Pseudogene entstehen, wenn Mutationen den Promotor inaktiviert haben (oder allgemeiner: wenn regulatorisch wichtige Sequenzen, welche die Aktivität des Gens steuern, zerstört wurden) oder wenn die Leseraster unterbrochen wurden (so dass resultierende Proteinsequenz erheblich verändert wurde) oder aber, wenn ganze Bereiche deletiert wurden (d.h. verloren gegangen sind). Unser Genom enthält etwa 20.000 Pseudogene, nur etwa 10% davon werden auch transkribiert. Ein extremes Beispiel ist das Gen RPL21, das für ein Ribosomalprotein kodiert. Von diesem Gen existieren inzwischen mehr als 140 Kopien in Form von Pseudogenen.
Oftmals sind sie aus Duplikationen hervorgegangen. Wenn nach einem solchen Ereignis eine der beiden Kopien beschädigt wird, hat dies keine negativen Auswirkungen, weil die andere ja noch vorhanden und intakt ist. Oft werden aber auch einzelne Gene durch Mutation zu inaktiven / funktionslosen Pseudogenen; die betreffende Funktion geht dann verloren. Aus dem menschlichen Genom sind viele solcher Pseudogene bekannt, die bei anderen Säugern funktional sind. Ein Beispiel ist die zur Synthese von Vitamin C nötige L-Gulono-γ-lacton-Oxidase, die bei Primaten, Meerschweinchen und Flughunden beschädigt ist, daher als Pseudogen vorliegt, während sie bei allen anderen Säugern intakt ist (YANG 2013). Konsequenterweise waren es in diesen drei Linien auch jeweils andere Mutationen, die das Gen funktionslos gemacht haben. Dies ist auf dem Boden der Evolutionstheorie vorhersagbar, denn die Wahrscheinlichkeit, dass voneinander unabhängige Verlustereignisse zufällig auf exakt denselben Mutationen beruhen, ist extrem gering (s. YANG 2013).

Eine besondere Klasse sind die sog. prozessierten Pseudogene; sie entstehen, wenn mRNA durch Reverse Transkriptase viralen Ursprungs (s.o.) in DNA „rückübersetzt“ wird (diese DNA nennt man dann cDNA) und danach im Rahmen von zellulären DNA-Reparaturmaßnahmen ins Genom eingebaut wird. Die Struktur solcher prozessierter Pseudogene ist ganz charakteristisch: Sie haben keine Introne mehr (weil die beim Spleißen zuvor entfernt worden waren) und keine Promotorelemente (weil die Promotoren von mRNA-kodierenden Genen nicht mit transkribiert werden), dafür aber einen mehr oder weniger langen polyAAbschnitt, weil ein solcher ans Ende jeder mRNA angefügt wird.4 Einen Überblick über die Evolution von Pseudogenen und dabei auch Beispiele für „funktionale Pseudogene“ liefern LI et al. (2013) sowie KORRODI-GREGÓRIO et al. (2013).
Der Begriff „Junk“-DNA wurde später auch auf andere Genombestandteile angewendet, die augenscheinlich funktionslos sind, also neben den erwähnten Pseudogenen z. B. auch für:
- Introns (also nicht-proteinkodierende Abschnitte, welche die Leserahmen unterbrechen),
- die meisten repetitiven Elemente („Repeats“ sind Sequenzen, die mehrfach bis vielfach - z.T. millionenfach – im Genom vorkommen: s.u.),
- Transposons (mobile genetische Elemente, die sich selbständig aus dem Genom ausschneiden und an irgendeiner anderen Stelle wieder einfügen können) sowie endogene Retroviren, die rund 8% des menschlichen Erbguts ausmachen (s.u.; vgl. auch KERENG 2010).
Allgemein kann Junk-DNA aber - wie gesagt - keinesfalls gleichgesetzt werden mit „nicht kodierender DNA“, weil auch zahlreiche nicht-proteinkodierende Abschnitte wie z.B. Promotoren wichtige (etwa regulatorische oder strukturelle und dabei Sequenz-abhängige!) Funktionen tragen (Abb. 4). Interessant und brisant zugleich ist, dass augenscheinlich rund 70% des menschlichen Genoms aus solcher „Junk-DNA“ besteht. Vor allen Dingen war es eine große Überraschung, dass der Mensch trotz seiner Komplexität mit erstaunlich wenig Genen (etwa 25.000) auskommt (LANDER et al. 2001), während viel weniger komplex organisierte Tiere fast ebenso viele Gene besitzen, einige sogar deutlich mehr, wie z.B. die Ackerschmalwand Arabidopsis. Der überwiegende Teil der DNA scheint demnach keine biologische Funktion zu haben, und es ist offenbar nicht die pure Anzahl der Gene, die hier relevant ist, sondern deren Organisation und Regulation im Genom sowie ihre „interne Komplexität“ (also das Vorhandensein verschieden vieler Spleißvarianten“.[5]

Abbildung: Regulatorische Elemente, Junk und nicht kodierende DNA bilden teilweise überlappende Kategorien, die leider dazu beitragen, Verwirrung zu stiften: Da „Junk-DNA“ vom Begriff her Funktionslosigkeit bzw. allenfalls eine sequenzunabhängige Funktion impliziert, darf man regulatorische Sequenzen wie Promotoren grundsätzlich nicht als Junk bezeichnen (der blau-rot überlappende Bereich sollte also eigentlich nicht vorkommen). Aus historischen Gründen sind die Zuordnungen aber nicht immer eindeutig.
Leider ist das Bild in den letzten Jahren noch komplizierter geworden: Wie eingangs erwähnt werden ca. 10% der Pseudogene transkribiert. Gehören sie damit zum Junk oder nicht? Offenbar sind sie biologisch aktiv, aber dies allein ist kein gutes Kriterium. Wenn man in funktionalen Kategorien argumentiert, kann man jedenfalls definieren, dass all dies "echte Müll-DNA" ist, was ohne jede physiologische Auswirkung deletiert werden kann. Somit müsste man Pseudogene, die zu nutzlosen RNAs transkribiert werden, zum Müll zählen. Pseudogene hingegen, deren Transkripte - wenngleich sie kein Protein mehr kodieren (können) - eine regulative Funktion erfüllen, muss man dann konsequenterweise als funktionale DNA betrachten (und noch nicht einmal als Junk, weil ihre regulatorische Funktion von der Sequenz abhängt).
Dies alles widerlegt zum einen den strengen „Adaptationismus“. Dieser betont die natürliche Selektion als treibende Kraft der Evolution: Mutationen, die einen Vorteil haben, werden rasch durch natürliche Selektion fixiert, wogegen neutrale oder gar negative Veränderungen aus der Population eliminiert werden. Bezogen auf funktionslose DNA bedeutet dies, dass sie einen „Kostenfaktor“ darstellt (bei jeder Zellteilung muss sie verdoppelt werden) und ihr Anteil durch deletierende Mutationen „klein gehalten“ wird. Mit anderen Worten, die streng adaptionistische Sichtweise erfordert ein „stromlinienförmiges“ Genom, mit einem geringen Anteil funktionsloser DNA, oder eine Funktion dieser DNA, die z.B. unabhängig von der konkreten Sequenz ist. Aber die Tatsache, dass es keinen (bekannten) Mechanismus gibt, der ständig und effektiv funktionslose Gene beseitigt (bzw. so rasch wie Pseudogene neu entstehen), stützt die Neutrale Theorie, nicht den strengen Adaptationismus. Sie bringt aber auch die Vertreter eines „intelligenten Designs“ in die Bredouille, die schon immer „gewusst“ haben, dass der intelligente Schöpfer seine Wesen rationell und sinnvoll gestaltet – was mit der Realität nun einmal nicht übereinstimmt.
7.2. Repeats
Mit „Repeat“ bezeichnet man in der Genetik die Wiederholung von Basenpaar Mustern in einem DNA- (oder RNA-)Strang. Es werden verschiedenste Klassen von Repeats unterschieden; die wichtigste Unterscheidung betrifft die relative Anordnung der Elemente zueinander (eine Auswahl z.B. bei: BURNS/BOEKE 2012; DEININGER 2011; GRANDI/AN 2013; KIM/MIRKIN 2013).
- Interspersed („eingestreute“) Repeats: Das betreffende DNA-Element sitzt in einzelnen Kopien verstreut im Genom. Hier werden unterschieden:
à LINEs (long interspersed nuclear elements). Es handelt sich hierbei um sog. Proviren: Retroviren (bekanntester Vertreter ist HIV) haben ein RNA-Genom, welches sie nach Infektion ihrer Wirtszelle in DNA umschreiben und dann als einige Kilobasen langes Provirus ins Genom der Zelle einbauen. Üblicherweise passiert das in Körperzellen, aber immer wieder einmal gelangen die so ins Genom eingebauten Viren auch in die Keimbahn, werden also weiter vererbt. Dass es sich hier – zunächst einmal! - nicht um „funktionale DNA“ handeln kann, ist unmittelbar einleuchtend und bedarf kaum einer Begründung: Viren sind Parasiten, der Organismus verfügt über ein großes Arsenal an Waffen, mit denen er sich gegen sie verteidigt. Die eingebrachten Gene gehören nicht zum Inventar der Zelle und erfüllen auch keine physiologischen Aufgaben. Im Gegenteil: Meist sind nur Teile von endogenen Retroviren aktiv, nur äußerst selten das ganze Provirus. Wenn ein ganzes Provirus aktiv ist, dann entstehen tausende von Viruspartikeln, und die betroffenen Zellen gehen zugrunde6 . Kein Wunder, dass ein Arsenal von Mechanismen evolvierte, um die Expression endogener Viren-DNA zu unterdrücken (LEUNG/LORINCZ 2012). Auch die Reverse Transkriptase (das Enzym, welches RNA in DNA rückschreibt) kann einigen Schaden anrichten (s.u.). Übrigens: einige dieser Elemente sind schon sehr alt, sie wurden ins Genom eingebaut, bevor sich die Entwicklungslinien von Mensch und Schimpanse trennten, so dass sie bis heute noch an einander entsprechenden Stellen im Genom sitzen.
à SINEs (short interspersed nuclear elements), z.B. die sog. Alu repeats („ALUs”), sind typischerweise 100–400 Basenpaare lange, häufig wiederholte und relativ frei im Genom verteilte DNA-Sequenzen. Die AluElemente entstammen dem 7-SL-RNA Gen, einem zellulären Gen, welches für einen Teil des sog. „signal recognition particle“ kodiert. Die Besonderheiten der Genstruktur bedingt, dass diese RNA – im Gegensatz zu proteinkodierenden mRNAs – ihren eigenen Promotor mit enthält. Wenn nun also in der Zelle durch ein integriertes Provirus Reverse Transcriptase vorhanden ist, so entsteht eine DNA-Kopie (eine sog. cDNA), die ggf. ins Genom integriert werden kann und dabei sofort wieder einen Promotor trägt, also transkribiert wird. Dies ist der Grund, dass sich dieses Element explosionsartig vermehren konnte: Primaten – und somit auch wir – tragen davon bis zu einer Million in ihrem Genom. ALUs sind bei heute aktiv, grob jeder 100ste Mensch trägt irgendwo in seinem Genom eine neue Kopie, wodurch sich zwei beliebige Menschen durch knapp 1000 Alu-Kopien (!) unterscheiden (XING et al. 2009). Es ist absurd zu postulieren, dass diese ALUs funktional sein sollen. Auch diese Familie ist – wie erwähnt – recht alt, daher gilt auch hier: Die Mehrzahl hat sich vor der Trennung von Mensch und Schimpansen ins Genom eingebaut, so dass sich die Evolutionsgeschichte auch anhand des Vergleichs der ALUs in den Genomen analysieren lässt.
à Tandem Repeats, die auch Satelliten genannt werden (der genaue Unterschied zwischen beiden Begriffen ist hier belanglos). Hier liegen die Wiederholungen auf dem Chromosom in direkter Folge hintereinander in mehreren bis Tausenden von Kopien. Unterschieden werden die Satelliten-Gruppen (nach der Länge der wiederholten Grundelemente) in Satelliten, Minisatelliten und Mikrosatelliten.
Da für diese Elemente – LINEs, ALUs, Pseudogene – die Entstehungsmechanismen bekannt sind (und beobachtet und nachvollzogen werden können; die Vorgängen sind dem Experiment zugänglich), ist evident, dass sie primär funktionslos sind. Aber Vorsicht! Die anfangs diskutierten Beispiele zeigen, dass man hier allzu schnell geneigt ist, mit menschlichen Maßstäben ans Thema heranzugehen. Machen wir uns klar: Die Evolution „weiß“ nicht, dass solche Elemente „eigentlich DNA-Müll“ sind, und dass sie diese darum „sinnvollerweise lieber beseitigen sollte“. Wenn solche Elemente im Genom sind, dann sind sie – ganz banal – zunächst einmal drin. Sofern sie tatsächlich funktionslos sind, unterliegen sie keinem Selektionsdruck. Sobald sie irgendeine Aktivität entwickeln oder auch nur irgendeine Wechselwirkung mit dem System – dem Organismus – besteht, entsteht ein Selektionsdruck. Und dabei ist es völlig gleichgültig, ob das betreffende DNA-Element zuvor „intakt“ oder „beschädigt“ oder „funktionslos“ war: Vorteilhaftes wird positiv selektiert, nachteiliges negativ.
Viele Pseudogene besitzen einen noch aktiven Promotor, sie werden also transkribiert; das Produkt ist aber funktionslos. Da die gebildete RNA derjenigen des Ursprungsgens ähnlich ist, kann es jedoch Interaktionen geben: Wenn die ursprüngliche Kopie intakt geblieben ist, kann die vom Pseudogen transkribierte RNA regulative Funktion übernehmen – und so ist es in einigen Fällen mittlerweile auch nachgewiesen (GERSTEIN/ZHENG 2007). Die einfache Frage, ob Pseudogene nun „Müll“ sind oder „funktional“, ist also differenziert zu beantworten: In den meisten Fällen ist es offensichtlich funktionsloser Ballast, in etlichen Fällen haben sie eine neue Funktion angenommen.
ALU-Repeats können erheblichen Schaden anrichten: Dadurch, dass sie in so hoher Anzahl vorhanden sind, können sie bei der DNA-Reparatur (FAZZA et. al. 2009, BELZIL 2007) oder während der Keimzellbildung die Rekombination der elterlichen Chromosomen stören, so dass es zu chromosomalen Veränderungen kommt. Durch ihre stetige Vermehrung und damit durch Integration ins Genom können sie Gene zerstören. Insbesondere können sie, wenn sie sich in Promotorbereiche hinein setzen, deren Aktivität verändern (JACOBSEN et al. 2009).
Nun hat sich während der Sequenzierung des Humangenoms etwas sehr interessantes gezeigt: Es hat vor wenigen Millionen Jahren einen erheblichen Aktivitätsschub der ALUs in unserem Genom gegeben, ablesbar an der Verteilung der Sequenzunterschiede zur Ursprungssequenz (s. Abb. 18 in LANDER et al. 2001). Berücksichtigt man zudem, dass etliche von ihnen in oder nahe bei Promotoren sitzen, wird klar, dass diese Periode hoher ALU-Aktivität Auswirkungen auf die Aktivität bzw. auf das Aktivitätsmuster menschlicher Gene gehabt hat. Es ist also sehr gut möglich, dass sie eine wichtige Rolle in unserer Evolution spielten.
Somit kann man wieder die eingangs gestellte Frage stellen: Sind ALUs denn nun „schädlich“, sind sie „parasitische DNA“ und somit „Feinde unseres Organismus“, oder waren sie „wichtige genetische Elemente“? Die Frage ist – so gestellt – sinnlos, denn es ist beides in gewissem Umfang zutreffend. Man kann es nur wiederholen: Erstens sind solch intentionale Ausdrücke unangemessen, weil sie die Natur des Evolutionsprozesses nicht angemessen widerspiegeln, sondern verzerren: Begriffe wie „parasitisch“ und „Feinde“ sind menschliche Kategorien, bei deren Anwendung auf die Natur man sehr vorsichtig sein muss. Zweitens ist ein „Entweder-Oder“ unangebracht – es kommt, so wie beim Gras und beim Weidevieh, immer wieder vor, dass beides gleichzeitig gilt. Wenn Lebewesen evolviert sind und sich in dieser Hinsicht grundlegend von technischen Apparaten unterschieden, so würde man auf dem Boden der Evolutionstheorie auch nichts anderes erwarten. Aber schauen wir uns schlussendlich die Gruppe der Tandem-Repeats noch etwas näher an.
7.3. Tandem repeats
Tandem repeats sind repetitive DNA-Elemente, die als solche Wiederholungen immer derselben Nukleotidsequenzen direkt hintereinander darstellen. Das Grundmuster kann von Fall zu Fall auch in leicht abgewandelter Form auftreten, wobei die jeweiligen Repeats umso häufiger im Genom vorkommen, je kürzer die Repeat-Elemente (also die „Wiederholungsgrundmuster“) sind. Ein Beispiel für ein trimeres (das heißt aus 3 Basenpaaren bestehendes) Tandem repeat wäre etwa die Sequenz „TACTACTACTAC“, in der die drei Nukleotidbasen Thymin, Adenin und Cytosin vier Mal auftreten bzw. repetiert werden. Der Anteil solch repetitiver DNAs im Genom der Säugetiere ist erstaunlich hoch. Etwa 50% der menschlichen DNA ist mittel- bis hochrepetitiv, besteht also aus den verschiedenen Repeat-Klassen.
7.4. Tandem repeats: Strukturen mit einer Funktion?
Tatsächlich haben einige wenige repetitive DNAs sehr wohl eine sequenzabhängige Funktion, darunter auch Protein-kodierende Gene. Das Kollagen-Gen sowie die strukturell wichtigen (nicht-kodierenden) Repeats im Zentromer und an den Telomeren sind Beispiele dafür, und es gibt noch etliche andere Elemente im Genom, die repetitive Sequenzabschnitte aufweisen. Eines der eindrucksvollsten Beispiele eines kodierenden Repeats findet sich im Kollagen-Gen, dessen Struktur wohl mit die am meisten ausgedehnten repeats aufweist, die man heute in Protein-kodierenden Bereichen kennt (MATSUSHIMA et al. 2009). Im Prinzip bestehen die kodierenden Bereiche dieses Gens (die so genannten Exons) aus der sechsfachen Wiederholung immer derselben 9-bp-Grundsequenz, die für das Aminosäurentriplett Gly-Pro-Pro kodiert, also insgesamt aus 54 Basenpaaren bzw. einem Vielfachen davon (108 bp). Unzählige dieser 54-bp- oder 108 bpExons bilden in ihrer Gesamtheit das Kollagen-Gen. Infolge von Genmutation variiert die Abfolge zwar leicht, aber das repetierte Grundmuster ist signifikant und lässt sich in der Aminosäuresequenz deutlich erkennen (s. Abb. 5).
An der Struktur repetitiver DNA lässt sich oft deren Entstehungsmechanismus erkennen: Solche repeats entstehen durch illegitime crossover (oder andere Duplikationsereignisse), helical slippage7 während der Replikation sowie durch Verschmelzung unterschiedlicher Genbereiche (Domain shuffling).

Abbildung 5: Ausschnitt aus dem Gen, welches für das Kollagen kodiert. Oben ist ein Exon abgebildet, welches aus 54 Basenpaaren (bp) besteht. Darin sticht die 9-bp-Grundsequenz hervor, die für die drei Aminosäuren „Glycin- Prolin-Prolin“ kodiert. Dieses tritt in vielfältiger Variation mehrmals in Erscheinung. Das Gen besteht als Ganzes wiederum aus zahlreichen solcher 54-bp-Exons oder Vielfachen davon – ein klarer Hinweis auf seinen evolutionären Ursprung durch zahlreiche Tandem-Duplikationen in der Vergangenheit.
Da es sich bei den Mechanismen der Genduplikation und Genfusion um theoretisch verstandene und experimentell nachgewiesene Mechanismen handelt, ist im Fall des Kollagens folgendes Szenario wahrscheinlich: Die Nona-RepeatGrundeinheit entstand mehr oder weniger zufällig und wurde durch einen der genannten Mechanismen (Tandemduplikation) zu einem kurzen Repeat vervielfältigt. Daraufhin geriet es, wie ungezählte andere DNA-Abschnitte in der Evolution immer und immer wieder auch, unter die Kontrolle eines Promotors. Dieses Repeat kodierte nun für ein kurzes Protein mit repetitiven Tripeptid-Einheiten, welches die physikochemisch hochinteressante Eigenschaft hat, Triple-Helices bilden zu können. Daraufhin schloss sich eine Serie von Tandemduplikationen an, was den kodierenden Bereich entsprechend verlängerte. Da dieses Protein zufällig mit einer Signalsequenz verschmolzen war oder einem anderen extrazellulären Protein anfusioniert wurde, landete es im Extrazellularraum – dort bildete es Filamente, die die Matrix verstärken. Damit haben wir eine sequenzabhängige Funktion und einen Selektionsdruck, der das Repeat zwar modifizieren kann (Länge, Feinheiten der Zusammensetzung), als solches aber konservieren muss. Weitere Serien von Tandemduplikationen sowie auch Deletionen und Domain Shuffling Ereignissen modifizierten die Kollagen-Gene, so dass sie heute eine ganze Genfamilie bilden.
Das Kollagen-Gen ist also einer der eher seltenen Fälle eines für ein Protein kodierenden Repeats (MATSUSHIMA et al. 2009). Deren Persistenz im Genom ist ganz einfach durch Selektion zu erklären: Im Kollagen ist diese Dreierabfolge nötig, damit sich die typisch verdrillte, fadenförmige Molekülgestalt des Strukturproteins Kollagen in Sehnen und Bändern ausbilden kann. Somit sind nur wenige - und ganz bestimmte – Varianten möglich, die repetitive Struktur bleibt durch Selektion erhalten. Die meisten Repeats aber kodieren weder für ein Protein, noch für eine funktionale RNA. Ansonsten sind nur noch Zentromer- und Telomer-Repeats – sie sind wichtig für die Chromosomen-Struktur – prominente Beispiele für funktionale tandem-repetitive DNA.
Weshalb aber verbleiben auch die übrigen, nicht kodierenden und offenbar auch anderweitig funktionslosen „tandem repeats“ im Genom? Hier ist es die Dynamik des Genoms, die für ihr Wachsen und Schrumpfen sorgt. Sobald per Zufall in nicht-kodierender DNA ein kurzes Repeat entstanden ist, kann es durch sog. „unequal“ oder auch „illegitimes“ Crossover anwachsen. Ein weiterer Mechanismus ist die sog. helical slippage: Bei der DNA-Replikation öffnen sich die DNAEnden aus thermodynamischen Gründen immer wieder, um sich praktisch sofort wieder zu schließen. Wenn nun ein Repeat vorliegt, dann kann es dabei zu einem Verrutschen kommen, wodurch sich die Anzahl der Repeat-Elemente erhört. Dies liegt schlicht am Mechanismus der DNA-Replikation und -reparatur; funktionale Gründe liegen nicht vor. Wenn nun diese Repeats funktionslos sind, dann bringen sie weder Vor- noch Nachteile mit sich. Ergo entscheidet lediglich die Dynamik (bzw. die Statistik) der betreffenden Vorgänge, mit welcher Häufigkeit Repeats entstehen, wachsen, schrumpfen, verschwinden.
Um abermals Missverständnissen vorzubeugen: In dem Moment, da DNA Elemente - welche auch immer - entstanden sind, sind sie im Genom vorhanden und unterliegen einem Selektionsdruck, abhängig von ihrer Auswirkung. Sie mögen Genstrukturen und Splicing-Muster, Promotoraktivitäten und Chromosomenstrukturen, Rekombinationsraten und Genomstabilität beeinflussen oder auch nicht - je nach Art, Ort und Anzahl. Der Einfluss mag vor- oder nachteilig sein, je nach Auswirkung. Insofern ist die Frage nach „Funktion“ komplex und die Frage nach „Sinn“ eine sinnlose (TOMILIN 2008; GEMAYEL et al. 2010; PLOHL et al. 2008).
8. Das ENCODE-Projekt: Hat 80% der (Junk-) DNA eine Funktion?
Vor einigen Monaten geisterten Nachrichten durch die Presse, die plötzlich alles infrage stellten. In zahlreichen populären Darstellungen heißt es, die Existenz von „Junk-DNA“ sei im Rahmen neuer Studien als Mythos entlarvt worden. Diese DNA sei, entgegen der Meinung der Evolutionsbiologie, keineswegs funktionsloser Müll, denn 80% der DNA erfülle „wichtige Aufgaben“ (ENCODE 2012; DE SOUZA 2012; QU/FANG 2013). Dies hat vor allem im Lager der Schöpfungsanhänger wahre Begeisterungsstürme ausgelöst und zu der überzogenen Behauptung geführt, das Junk-DNA-Konzept gehöre nun der Geschichte an (z.B. LUSKIN 2012; TYLER 2012; WORT UND WISSEN 2012). Solchen Behauptungen verleiht TYLER (2012) durch einen eigens illustrierten Grabstein Nachdruck (Abb. 6).

Abbildung 6: Das ENCODE-Projekt und das vermeintliche Ende des JunkDNA-Konzepts. Oder leben Todgesagte doch länger als erwartet? Aus TYLER (2012).
Dabei wurde vor allem eines deutlich – die biologische Unkenntnis derer, die solche Nachrichten unreflektiert aufschnappten und in einen Beleg für ein intelligentes Design umzumünzen versuchten. So amüsant TYLERs Polemik ist, so absurd ist sie auch. Behauptungen, wie dass fast alle Genom-Bereiche eine Funktion hätten, blenden kurzerhand sowohl die empirischen Belege, die dagegen sprechen, als auch die hochkarätig publizierten Kritiken am ENCODE-Projekt aus. Aber der Reihe nach: Was ist ENCODE und was wurde im Rahmen des Projekts eigentlich gezeigt?
ENCODE (das Akronym für: Encyclopedia of DNA Elements) ist ein Forschungsprojekt, das 2003 vom US-amerikanischen National Human Genome Research Institute initiiert wurde. Sein Ziel besteht darin, alle funktionellen Elemente des menschlichen Genoms zu identifizieren und das sog. Transkriptom zu charakterisieren. Durchgeführt wurde das Projekt von einem Forschungskonsortium, dem etwa 30 Arbeitsgruppen an verschiedenen Instituten angehören (DE SOUZA 2012; QU/FANG 2013). Im September 2012 erschien in der Fachzeitschrift „Science“ ein Artikel von Elizabeth PENNISI, worin es heißt, ENCODE habe nachgewiesen, dass 80% des menschlichen Genoms biochemische Aufgaben erfüllten. Diese Einsicht sei, so PENNISI weiter, der „Todesstoß für die Idee, dass unsere DNA überwiegend mit nutzlosen Basenpaaren angefüllt ist“ (PENNISI 2012).
Das Medienecho dieser reißerischen Schlagzeile war groß. Im „Scientific American“ etwa heißt es: „... der größte Teil des genetischen Codes zwischen den Genen kontrolliert wichtige Funktionen von Leben und Gesundheit.“ (Übersetzung: Die Autoren.) Dabei fiel kaum jemandem auf, was sich aus evolutionsbiologischer Sicht tatsächlich über Junk-DNA aussagen lässt und welch irreführenden Funktionsbegriff ENCODE ihren Studien zugrunde legte.
Um den Sachverhalt ins rechte Licht zu rücken: Die Messlatte für „Funktion“ wurde vom ENCODE-Projekt äußerst niedrig gehängt, wobei alles, wofür eine Funktion nicht strikt ausgeschlossen werden konnte, als „potenziell funktional“ deklariert wird. Insbesondere wurden DNA-Abschnitte schon als funktional erachtet, wenn diese nur in irgendeiner (wenn auch nicht unbedingt sinnvollen) Art und Weise aktiv sind. Wird etwa die DNA eines defekten Gens zu unnützer „Junk-RNA“ umgeschrieben, ist dies nach den ENCODE-Maßstäben bereits eine Funktion! Folglich wird jedes Stückchen DNA identifiziert, für das auch nur die geringste Chance besteht, dass es eine Funktion tragen könnte. Selbstverständlich kann man so vorgehen, um später die Funktionen nachzuweisen und zu ergründen – dieser Schritt hätte aber erst erfolgen müssen, und das ist im Presserummel um die ENCODE-Meldung komplett untergegangen. Aus diesem Grund wird das Projekt – genauer: der Tenor der ENCODE-Publikationen – von vielen Seiten heftig kritisiert. Eine kleine Auswahl dazu: TIMMER (2012); EDDY (2012); EDDY (2013); DOOLITTLE (2013); GRAUR et al. (2013). Insbesondere in der öffentlichen Wahrnehmung gehen „Funktion nicht mit Sicherheit auszuschließen“ und „(wichtige) Funktion nachgewiesen“ komplett durcheinander.
Bei alledem wird völlig übersehen, dass kein Biologe in den letzten 30 Jahren alle nicht-protein-kodierenden DNA-Bereiche zu „funktionslosem Müll“ deklarierte. Abschnitte, die Gene steuern, sind schon lange bekannt. Schon gar nicht wird behauptet, Junk-DNA entfalte keine wie auch immer geartete molekulargenetische Aktivität (also „Funktion“ im weitesten Sinne) – sie erfüllt, wie oben dargelegt, nur eben meist keine [strikt] sequenzabhängige Funktion – falls überhaupt.
Kürzlich widerlegte ein Forscherteam um Luis HERRERA-ESTRELLA vom Laboratorio Nacional de Genómica para la Biodiversidad (Langebio) in Mexiko und Victor ALBERT von der University at Buffalo die Vorstellung, dass Junk-DNA für komplexes Leben wichtige Funktionen erfülle. Den Beleg dafür, dass der größte Teil der Junk-DNA verzichtbar ist, liefert die fleischfressende Wasserpflanze Utricularia gibba (Zwerg-Wasserschlauch, Abb. 7). Sie hat den Großteil dessen eliminiert, was normalerweise das Genom von Pflanzen ausmacht, nur noch 3% ihrer DNA bestehen aus Junk-DNA. Das bedeutet, dass auch ohne Junk-DNA eine voll funktionstüchtige, mehrzellige Pflanze überleben kann, die viele unterschiedliche Zellen, Organe, Gewebetypen und Blüten besitzt (IBARRA-LACLETTE et al. 2013).

Abbildung:
Die fleischfressende Pflanze Utricularia gibba entledigte sich des größten Teils dessen, was das Genom von Pflanzen ausmacht. Dies legt den Schluss nahe, dass der
größte Teil des Genoms höher entwickelter Lebensformen keine wichtige Funktion hat. © Alex POPOVKIN, lizensiert unter Creative Commons Namensnennung 2.0.
Quelle: www.flickr.com/photos/plants_of_russian_in_brazil/6754403777/
Für die Entbehrlichkeit eines Großteils nicht-kodierender DNA spricht auch das Ergebnis einer Studie, bei der zwei längere Abschnitte nichtkodierender DNA aus dem Genom von Mäusen entfernt wurden, was zu keinen merkbaren Unterschieden im Phänotyp führte (NÓBREGA et al. 2004). Solche Ergebnisse wurden von kreationistischen Autoren wie TYLER (2012) entweder komplett übersehen oder aber spezifisch ausgeblendet.
Ein weiterer Aspekt betrifft das sog. c-Wert Paradoxon (EDDY 2012; DOOLITTLE 2013 und die darin zitierten Publikationen): Der Komplexitätslevel eines Organismus verlangt eine gewisse Mindestgröße des Genoms, die bei komplexen Tieren größer ist als bei einfach strukturierten, bei letzteren jedoch immer noch größer ist als bei einzelligen Eukaryonten und bei diesen wiederum größer ist als bei Bakterien. Frei lebende Organismen brauchen ein größeres Genom als Parasiten, die viele ihrer physiologischen Funktionen über Bord geworfen haben usw. Umgekehrt jedoch korreliert die Genomgröße nicht mit der Komplexität der Lebewesen, hier gibt es keinen strikten Zusammenhang: Manche Fische haben ein dutzendfach größeres Genom als Säuger (uns inklusive); manche Einzeller (!) ein hundertfach größeres. Allein deswegen ist es schlicht unmöglich, dass überall in dieser gigantischen DNA-Menge Funktionalität herrscht. An diesen Erkenntnissen vermochten die Studien von ENCODE nicht zu rütteln, und es mehren sich die Wissenschaftler, die an der Interpretation der Studien von ENCODE massiv Kritik üben (s.o.). Die Identifikations-Parameter wurden allzu großzügig eingestellt.
Wie oben ausgeführt, ist aus evolutionärer Sicht die Herkunft und Existenz von Junk-DNA einfach zu erklären, aus Sicht der Neutralen Theorie wurde deren Existenz sogar vorausgesagt8 . Wie sich durch die Sequenzierungsprojekte ganzer Genome in den letzten Jahren herausgestellt hat, überschätzte OHNO zwar den Anteil, den Pseudogene am Genom einnehmen. Die meisten Pseudogene sind aber tatsächlich funktionslos im strengeren Sinne, d. h. ihre An- oder Abwesenheit macht keinerlei Unterschied aus. Mehr zu Pseudogenen findet man beispielsweise im TalkOrigins-Archive: Plagiarized Errors and Molecular Genetics.
9. Und was ist mit Intelligent Design?
Die ID-Anhänger freilich haben schon immer „gewusst“, dass Junk-DNA (zumindest teilweise) eine Funktion erfülle. Ja, die Funktionalität der vermeintlichen Junk-DNA sei aus Sicht der „Intelligent-Design-Theorie“ regelrecht vorhergesagt worden. Die Erwartungen „der Evolutionstheorie” [sic!] hätten sich dagegen nicht bestätigt. Bei MEYER (2004) lesen wir:
„Thus, ID says that we would expect to find function for junk-DNA because, in our experience, designers make things for a purpose. This leads to a POSITIVE expectation that junk-DNA will have function. Neo-Darwinism can do whatever it wants; ID has long-predicted that junk-DNA has function, and ID was right.“[9]
LUSKIN wiederum versichert uns (zitiert nach SCHU 2007):
„Intelligent design begins by studying the types of complexity produced by intelligent agents. We observe that intelligent agents produce things for a purpose, that is, to fulfill some function. This leads ID proponents to an expectation—yes, a prediction— that DNA will not tend to contain meaningless junk but will contain structures that have (or once had) a function for the organism. ID does not lead us to the expectation that our cells' DNA will be largely non-functional garbage.“[10]
Was die angeblich spezifische Voraussage von Intelligent-Design anbelangt, so fragt man sich, weshalb nach LUSKINs Ansicht nicht nur funktionale DNA, sondern auch DNA-Elemente, die „once had (!) a function for the organism“, im Genom des Menschen vorkommen (LUSKIN 2007). Darunter fallen funktionslose Pseudogene, für die OHNO (1972) den Begriff „Junk-DNA“ prägte. Ist dies ein „goalpostmoving“ 11? So wie die alles andere als spezifische Aussage, wieviel nicht-funktionale DNA im Genom ein Designer denn nun seiner Meinung nach da „purposeful“ abgelegt hat? Um die ISCID zu zitieren:
„An intelligent designer need not produce perfect designs - nor even functional designs.“[12]
Was also bleibt von der „spezifischen Voraussage“ des Intelligenten Designs noch übrig, wenn man die sich gegenseitig widersprechenden Aussagen von Intelligent-Design-Vertretern gegenüber stellt?
Um es kurz zu machen: Da ID-Anhänger behaupten, sie hätte gar keinen bestimmten Designer im Sinn, können sie logischerweise auch keine Aussagen bzw. Vorhersagen über seine Intentionen und Vorgehensweisen machen! Dementsprechend können sie auch nicht voraussagen, ob der oder die (?) Designer nicht auch unnötigen Firlefanz eingebaut oder einfach schlechtes Design produziert haben – und ob tatsächlich alles (oder das Meiste) eine Funktion haben muss.
Auch aus Sicht des biblischen Schöpfungsmythos, wonach Gottes Wege „unergründlich“ sind, lässt sich alles Mögliche – und das genaue Gegenteil – schlussfolgern. „Design“ ist nicht einmal semantisch klar definiert – niemand weiß, was „Design“ im Zusammenhang mit der Erschaffung der Arten konkret bedeuten soll. „Intelligent Design“ ist, um es in den Worten von KUMMER (2009, 153) zu formulieren, ein „metaphysischer Joker ohne Erklärungswert“, der sich schlicht überall einsetzen lässt. Eben weil mangels Kenntnis des postulierten Designvorgangs und dessen Randbedingungen keine konkreten, spezifischen Vorhersagen möglich sind, ist Design grundsätzlich unprüfbar und ein „Passepartout“, das pauschal die lästige Frage nach der Genese von Biosystemen abnimmt, den Sachverhalt aber letztlich verunklart statt erhellt.
10. Zusammenfassung
Wie wir gesehen haben, ist vor allem die Begriffsverwirrung um „Funktion“, „Nutzen“, „DNA-Junk“ und „DNA-Müll“ verantwortlich dafür, dass die Diskussion um die Funktion genomischer Elemente immer wieder aus dem Ruder läuft - selbst unter Fachleuten. Betrachtet man die Elemente unter funktionalen Aspekten, so findet man:
- die "open reading frames" in Exonen: Das sind Bereiche, die für Proteine kodieren - sie tragen sequenzabhängige Information, die im genetischen Code abgelegt ist.
- Transkribierte Elemente, die zwar für keine Proteine kodieren, aber (sequenzabhängig) funktional sind: Sie üben regulatorische oder anderweitige Funktion aus. Hierunter fallen einige Pseudogene, tRNA- und rRNA-Gene, viele kleine RNAs usw.
- Nicht-transkribierte Elemente, die sequenzabhängig strukturelle oder regulatorische Funktion tragen. Hierunter fallen regulatorische Bereiche wie Promotoren, Terminatoren, Enhancer, Silencer, Centromer- und Telomerbereiche usw.
- Nicht-transkribierte Elemente, die sequenzunabhängige, strukturelle (selten auch regulatorische) Funktion tragen. Dies wären dann Introne, die differenziell gespleißt werden oder Spacer, chromosomale „Abstandhalter“, die aus strukturellen Gründen in der richtigen Länge an der richtigen Stelle sitzen müssen. Wenn wir Junk-DNA als „DNA mit keinerlei Sequenzabhängiger Funktion“ definieren, so ist dies Junk-DNA, aber mitnichten funktionslos.
All diese Elemente haben gemein, dass sie einem positiven Selektionsdruck unterliegen, eine Deletion oder eine signifikante Änderung durch Mutationen ist für den Organismus nachteilig.
- Dann gibt es DNA-Bereiche, die ohne jede Folge frei mutieren oder deletiert werden können. Das wäre Junk-DNA und darüber hinaus funktionsloser Junk, also DNA-Müll im engeren Sinne. Das bedeutet aber nicht, dass diese Bereiche keinerlei Aktivität zeigen: Sie können sehr wohl zu nutzloser RNA transkribiert werden. Und damit können sie in der späteren phylogenetischen Historie eine Funktion annehmen.
- Und letztlich: Selbst Elemente, die als solche definitiv funktionslos sind, können in der Evolution eine wichtige Rolle gespielt haben, z.B. indem sie - wie die ALU-Repeats in der menschlichen Evolution - durch ihr Springen Promotor-Aktivitäten verändert haben. Womit sich der Kreis zum Anfang des Beitrags schließt: Freunde oder Feinde? Nutzlos oder wichtig? ...
Nach Abwägung aller Fakten lässt sich also festhalten: Die Evolutionstheorie kann die Herkunft sowie die Persistenz von Junk-DNA im Genom mechanismisch gut erklären; deren Existenz ist im Rahmen der „Neutralen Theorie“ vorhergesagt worden. Dagegen liefert „Intelligent Design“ keinen Beitrag zur wissenschaftlichen Diskussion, geschweige denn konkrete Vorhersagen über Existenz, Herkunft und Ausmaß der Junk-DNA.
Einzelnachweise
[1] Aus Sicht eines Ingenieurs ergibt das alles keinen Sinn – niemand kann behaupten, Ingenieure seien darauf angewiesen, ursprüngliche Organisationsmuster beizubehalten. Hier offenbart sich der Gegensatz zwischen Natur und Technik, Lebewesen und Artefakt: Artefakte werden einfach zusammen geschraubt. Solange sie nicht fertig sind, können Bauteile beliebig ausgetauscht, auf Teile, die in anderen Grundbauplänen eine tragende Rolle spielen, kann verzichtet werden. Lebewesen hingegen sind in jedem Stadium ihrer Entwicklung voll funktionsfähig (d.h. sie sind schon immer „fertig“), woraus sich aus evolutionärer Sicht die Notwendigkeit des Beibehaltens von Teilen der ursprünglichen Entwicklungsprogramme ableitet. Deshalb zählen transitorische Merkmale, die aufgrund ihres Baus noch ihre Primärfunktion verraten, diese inzwischen aber komplett verloren haben, zu den eindrucksvollsten Belege für eine stammesgeschichtliche Entwicklung, auch wenn den betreffenden Strukturen inzwischen eine sekundäre Funktion oktroyiert wurde.
[2] Dies sind relativ neu entdeckte Arten nicht-proteinkodierender RNAs: Also RNAMoleküle, die als solche (also als RNA) eine Funktion ausüben, ohne dass sie in Proteine translatiert werden.
[3] Das sind Sequenzelemente, welche insbesondere die Aktivität von Genen, und dabei auch die gegenseitige Abstimmung der Genaktivitäten zueinander regulieren.
[4] Diese Zusammenhänge sind komplex – in wenigen Worten: Ein proteinkodierendes Gen enthält notwendigerweise den Bereich, der tatsächlich für das betreffende Protein kodiert: den sog. Leserahmen. Daneben enthält es vor dem Leserahmen Steuerungselemente, den sog. Promotor. Bei Pflanzen und Tieren ist der Leserahmen zusätzlich meistens noch durch nicht-kodierende Abschnitte, die sog. Introne, unterbrochen. Der Promotor wird nicht in mRNA transkribiert, die Introne werden unmittelbar nach der Transkription entfernt (das sog. Splicen). Ergo kann ein aus mRNA in cDNA rückgeschriebenes Gen weder einen Promotor, noch Introne enthalten.
[5] Konsequenterweise gibt es für jede Organisationsstufe (oder –höhe) eine minimale Genomgröße, also eine ungefähre Anzahl von Genen, derer es bedarf, um ein entsprechend komplexes Lebewesen zu kodieren: Unter einem Megabasenpaar (MBp) für parasitische Bakterien, einige MBp für Bakterien, ein Dutzend MBp für einzellige Eukaryonten, 100 MBp für mehrzellige Tiere und Pflanzen. Interessanterweise gibt es aber keine Obergrenze, die mit der Komplexität der betreffenden Organismen korreliert wäre – dieses Faktum nennt man das „C-Wert-Paradox“. So hat Amoeba dubia, eine einzellige Amöbe, ein Genom von 670.000 MBp, das ist 200mal größer als unser Genom. In solchen Fällen lässt sich die Genomgröße durch nicht-funktionale „Junk“-DNA erklären, nicht aber durch physiologische, biochemische oder entwicklungsbiologische Notwendigkeit.
[6] Auf diese Weise schädigt das HI-Virus die sog. T-Helferzellen (wichtige Zellen des Immunsystems)
[7] Ein „Verrutschen“ der kopierenden Polymerase um eine oder mehrere Repeat-Einheiten. Dies kann in vitro, also im Labor, nachgemacht und untersucht werden.
[8] OHNO: “The points I wish to make are: 1) Natural selection is an extremely conservative force. So long as a particular function is assigned to a single gene locus in the genome, natural selection only permits trivial mutations of that locus to accompany evolution. 2) Only a redundant copy of a gene can escape from natural selection and while being ignored by natural selection can accumulate meaningful mutation to emerge as a new gene locus with a new function. Thus, evolution has been heavily dependent upon the mechanism of gene duplication. 3) The probability of a redundant copy of an old gene emerging as a new gene, however, is quite small. The more likely fate of a base sequence which is not policed by natural selection is to become degenerate. My estimate is that for every new gene locus created about 10 redundant copies must join the ranks of functionless DNA base sequence. 4) As a consequence, the mammalian genome is loaded with functionless DNA.” [Quelle: Ohno 1973, vollständige Literatur-Referenz bei Genomicron]
[9] „Daher sagt ID voraus, dass wir erwarten können, in Junk-DNA Funktionen zu finden, den nach unserer Erfahrung kreiert ein Designer Dinge zu einem bestimmten Zweck. Daraus können wir positiv die Erwartung formulieren, dass Junk-DNA eine Funktion hat. Der Neo-Darwinismus kann machen was er will; ID hat schon seit langem vorhergesagt, dass Junk-DNA eine Funktion hat und ID hat Recht behalten.“
[10] „Intelligent Design beginnt, indem verschiedenen Komplexitäts-Typen untersucht werden, die von unterschiedlichen intelligenten Akteuren produziert wurden. Wir beobachten, dass intelligente Akteure Dinge zu einem Zweck produzieren, also um eine Funktion zu erfüllen. Das bringt ID-Vertreter zu einer Erwartung, sogar zu einer Vorhersage: dass DNA prinzipiell zunächst einmal keinen bedeutungslosen „Schrott“ enthält, sondern Strukturen, die eine Funktion haben (oder einst hatten). Aufgrund von ID werden wir nicht erwarten können, dass ein Großteil der genomischen DNA funktionsloser Müll ist.“
[11] „Verschieben des Torpfostens“: Verändern der Spielregeln mitten im Spiel u. dgl.
[12] http://www.iscid.org/encyclopedia/Optimal_Design,_Argument_From, Zugriff 2.5.2013
Literaturverzeichnis
BELZIL, C. (2007) Alu Elements and Human Disease. Lethbridge Undergraduate Research Journal.
2(1)
BEYER, A. (2007) Was ist Wahrheit? - oder - Wie Kreationisten Fakten wahrnehmen und wiedergeben. In: Kutschera, U. (Hg.) Kreationismus in Deutschland: Fakten und Analysen. Lit-Verlag. ISBN
978-3-8258-9684-3
BOMMAS-EBERT, U./TEUBNER, P./VOß, R. (2006) Kurzlehrbuch Anatomie und Embryologie. 2. Auflage. Thieme-Verlag. ISBN 3131355328, S. 59ff.
BURNS, K.H./BOEKE, J.D. (2012) Human transposon tectonics. Cell 149(4), 740- 752. doi: 10.1016/j.cell.2012.04.019.
DAWKINS, R. (2009) History written all over us. In: The greatest show on Earth. Free Press. ISBN 978-1-4165-9478-9, 360–362.
DE SOUZA, N. (2012) The ENCODE project. Nat Methods 9(11),1046.
DEININGER, P. (2011) Alu elements: know the SINEs. Genome Biol. 12(12), 236. doi: 10.1186/gb-2011-12-12-236.
DOOLITTLE, W.F. (2013) Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A 110(14), 5294-5300. doi: 10.1073/pnas.1221376110.
EDDY, S.R. (2012) The C-value paradox, junk DNA and ENCODE. Curr Biol. 22(21), R898-R899. doi: 10.1016/j.cub.2012.10.002.
EDDY, S.R. (2013) The ENCODE project: missteps overshadowing a success. Curr Biol. 23(7),R259-R261. doi: 10.1016/j.cub.2013.03.023.
ENCODE Project Consortium/Bernstein, B.E./Birney, E./Dunham, I./Green, E.D./Gunter, C./Snyder, M. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414),57-74.
doi: 10.1038/nature11247.
FAZZA, A.C./SABINO, F.C./DE SETTA, N./BORDIN, N.A. Jr/DA SILVA, E.H./CARARETO, C.M. (2009) Estimating genomic instability mediated by ALU retroelements in breast cancer. Genet Mol Biol. 32(1),
25-31.
GEMAYEL, R./VINCES, M.D./LEGENDRE, M./VERSTREPEN, K.J. (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 44, 445-77. doi:
10.1146/annurev-genet-072610-155046
GENOMICRON www.genomicron.evolverzone.com/2012/09/encode-2012-vs-comings1972/ GERSTEIN, M./ZHENG, D. (2007) Das heimliche Wirken der Pseudogene. Spektrum der Wissenschaft 4/2007
GRANDI, F.C./AN, W. (2013) Non-LTR retrotransposons and microsatellites: Partners in genomic variation.Mob Genet Elements 3(4), e25674.
GRAUR, D./ZHENG, Y./PRICE, N./AZEVEDO, R.B./ZUFALL, R.A./ELHAIK, E. (2013) On the immortality of television sets: „function“ in the human genome according to the evolution-free gospel of ENCODE.
Genome Biol Evol. 5(3), 578- 590. doi: 10.1093/gbe/evt028.
IBARRA-LACLETTE, E. et al. (2013) Architecture and evolution of a minute plant genome. Nature 498, 94–98.
JACOBSEN, B.M./JAMBAL, P./SCHITTONE, S.A./HORWITZ, K.B. (2009) ALU repeats in promoters are position-dependent co-response elements (coRE) that enhance or repress transcription by dimeric and
monomeric progesterone receptors. Mol Endocrinol. 23(7), 989-1000.
KERENG (2010) Evolutionsbeweis durch endogene Retroviren. Zusammenfassung und Linkliste ausführliche Literatursammlung unter: http://kereng.wordpress.com/2009/11/18/Evolutionsbeweis-durch-ERV---
Zusammenfassung-und-Linkliste/
KHOSRAVI, A./MAZMANIAN, S.K. (2013) Disruption of the gut microbiome as a risk factor for microbial infections (Review). Curr Opin Microbiol. 16(2), 221- 227. doi:
10.1016/j.mib.2013.03.009.
KIM, J.C./MIRKIN, S. (2013) The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 23(3), 280-288. doi: 10.1016/j.gde.2013.04.009.
KLOWDEN, M.J. (2007) Physiological systems in insects. Elsevier/Academic Press, 497-499.
KORRODI-GREGÓRIO, L./ABRANTES, J./MULLER, T./MELO-FERREIRA, J./MARCUS, K./DA CRUZ, E./SILVA, O.A./FARDILHA, M./ESTEVES, P.J. (2013) Not so pseudo: the evolutionary history of protein phosphatase
1 regulatory subunit 2 and related pseudogenes. BMC Evol Biol. 13(1), 242.
KUMMER, C. (2009) Der Fall Darwin. Evolutionstheorie contra Schöpfungsglaube. Pattloch-Verlag.
LANDER, E.S. et al. (2001) International Human Genome Sequencing Consortium: 2001 Initial sequencing and analysis of the human genome. Nature 409(6822), 860-921.
LI, W./YANG, W./WANG, X.J. (2013) Pseudogenes: pseudo or real functional elements? J Genet Genomics 40(4), 171-177. doi: 10.1016/j.jgg.2013.03.003.
LEUNG, D.C.; LORINCZ, M.C. (2012) Silencing of endogenous retroviruses: when and why do histone marks predominate? Trends in biochemical sciences 37, 127–133.
LUSKIN, C. (2007) Richard Gallagher Frames Intelligent Design Proponents While Rewriting the History of Junk-DNA (Part 2). www.evolutionnews.org/2007/07/richard_gallagher_frames_intel_1003919.
html#more.
LUSKIN, C. (2012) Junk No More: ENCODE Project Nature Paper Finds „Biochemical Functions for 80% of the Genome“ www.evolutionnews.org/2012/09/junk_no_more_en_1064001.html. Zugr. a.
11.01.2014.
MAHNER, M. (2001) Funktion. In: Lexikon der Biologie, Bd. 6. Spektrum Akademischer Verlag, 110-111.
MAHNER, M./BUNGE, M. (2000) Philosophische Grundlagen der Biologie. SpringerVerlag.
MAHNER, M./BUNGE, M. (2001) Function and functionalism: A synthetic perspective. Philosophy of Science 68(1), 75-94.
MATSUSHIMA, N./TANAKA, T./KRETSINGER, R.H. (2009) Non-globular structures of tandem repeats in proteins. Protein Pept Lett. 16(11), 1297-1322.
MCNAMARA, J.M. (2013) Towards a richer evolutionary game theory. J R Soc Interface 10(88), 20130544. doi: 10.1098/rsif.2013.0544.
MEYER, S.C. (2004) The origin of biological information and the higher taxonomic categories. Proceedings of the Biological Society of Washington 117(2), 213-239.
NIH Human Microbiome Project www.human-microbiome.org/
NÓBREGA, M.A./ZHU, Y./PLAJZER-FRICK, I./AFZAL, V./RUBIN, E.M. (2004) Megabase deletions of gene deserts result in viable mice. Nature 431(7011), 988-993.
NUISMER, S.L./JORDANO, P./BASCOMPTE, J. (2013) Coevolution and the architecture of mutualistic networks. Evolution 67(2), 338-354. doi: 10.1111/j.1558- 5646.2012.01801.x.
OHNO, S. (1972) So much „junk” DNA in our genome. In: Smith, H.H. (Hg.) Evolution of Genetic Systems. Gordon and Breach, 366-370.
OHNO, S. (1973) Evolutionary reason for having so much junk DNA. In: Pfeiffer, R.A. (Hg.) Modern aspects of Cytogenetics in man. F. K. Schattauer.
PENNISI, E. (2012) ENCODE Project Writes Eulogy for Junk DNA. Science 337(6099), 1159-1161. DOI: 10.1126/science.337.6099.1159
PLOHL, M./LUCHETTI, A./MESTROVIĆ, N./MANTOVANI, B. (2008) Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric
(hetero)chromatin. Gene 409(1-2), 72-82. doi: 10.1016/j.gene.2007.11.013.
QU, H./FANG, X. (2013) A brief review on the Human Encyclopedia of DNA Elements (ENCODE) project. Genomics Proteomics Bioinformatics 11(3), 135- 141. doi: 10.1016/j.gpb.2013.05.001.
SCHU, S. (2007) Nachtrag: Casey Luskin „erklärt“, warum ID Funktionen in JunkDNA voraussagt. http://evilunderthesun.blogspot.de/2007/07/oh-wiekonnte-ich-das-bersehen.html
SUWEIS, S./SIMINI, F./BANAVAR, J.R./MARITAN, A. (2013) Emergence of structural and dynamical properties of ecological mutualistic networks. Nature 500(7463), 449-452. doi:
10.1038/nature12438.
THE INTERNATIONAL HUMAN MICROBIOME CONSORTIUM: http://www.hmpdacc.org/
TIMMER, J. (2012) Most of what you read was wrong: how press releases rewrote scientific history (Kommentar). Ars Technica 10.Sept. 2012
http://arstechnica.com/staff/2012/09/most-of-what-you-read-was-wronghow-press-releases-rewrote-scientific-history/2/. Zugr. a. 11.01.2014.
TOMILIN, N.V. (2008) Regulation of mammalian gene expression by retroelements and non-coding tandem repeats. Bioessays 30(4), 338-348. doi: 10.1002/bies.20741.
TYLER, D. (2012) Post details: ENCODE - The Junk DNA concept must be consigned to history. www.arn.org/blogs/index.php/literature/2012/09/10/encode_the_junk_dna _concept_must_be_cons. Zugr. a.
11.01.2014.
WORT UND WISSEN (2012) Mega-Datenbank zeigt: Fast alles Erbgut hat eine Aufgabe. www.facebook.com/permalink.php?id=156521214469175&story_fbid=1435 07799124215. Zugr. a. 11.01.2014.
XING, J./ZHANG, Y./HAN, K./SALEM, A.H./SEN, S.K./HUFF, C.D./ZHOU, Q./KIRKNESS, E.F./LEVY, S./BATZER, M.A./JORDE, L.B. (2009) Mobile elements create structural variation: analysis of a complete
human genome. Genome Res 19, 1516-1526.
YANG, H. (2013) Conserved or lost: molecular evolution of the key gene GULO in vertebrate vitamin C biosynthesis. Biochem Genet. 51(5-6), 413-425. doi: 10.1007/s10528-013-9574-0.
Gastbeitrag von Prof. Dr. Andreas Beyer und Dr. Martin Neukamm
Kommentar schreiben